400-029-0925
400-029-0925“儿童杀手”、“死亡恶魔”——弥漫性内生性脑桥胶质瘤(DIPG)究竟是什么?
弥漫性内生性脑桥胶质瘤(DIPG)是一种致命的恶性儿童肿瘤,在脑桥中弥漫生长。这种损害性疾病的诊断年龄中位数为6-7岁,很少在成人中发现。鉴于其生长位置处于脑干“手术禁区”,目前的治疗选择有限并且预后很差——只有不到10%的患者自诊断之日起存活超过2年。
DIPG占全部小儿脑肿瘤的80%,发生在脑干。在组织学上,这些肿瘤与间变性星形细胞瘤具有相同的特征(III级)或胶质母细胞瘤(GBM)(IV级)。国际卫生组织2016年脑肿瘤分类中,在组蛋白H3(3.1或3.3)中具有K27M突变的小儿神经胶质瘤,具有弥漫性生长模式中线位置称为弥漫性中线胶质瘤,H3K27M突变体;该定义也包括了DIPG带有K27M突变的病例。
近年来,DIPG治疗以及分子生物学取得了进展。以前,儿童高级别胶质瘤(HGG)被认为类似于成人HGG并进行临床治疗。然而,后来国内外专家认识到不同的分子改变将DIPG与其成人HGG对应物区分开来。
INC国际神经外科顾问团成员、加拿大SickKids儿童医院脑瘤研究中心主任、国际神经外科杂志《Journal of Neurosurgery》主编James T.Rutka教授在2023年“三届国际神经外科顾问团云端峰会”期间,对目前进行的DIPG研究进行了演讲交流,演讲议题为《Diffuse intrinsic pontine glioma–Phase I clinical trial using MR-guided focused ultrasound for drug delivery》(DIPG弥漫内生型脑桥胶质瘤-磁共振引导聚焦超声“磁波刀”治疗I期临床试验),交流了国际小儿神外专家James T.Rutka教授对磁波刀在胶质瘤的目前国际前沿研究。
而在Rutka教授参与撰写的《Diffuse intrinsic pontine glioma:current insights and future directions》论文中,对DIPG也进行了系统概述,包括DIPG表现、分型及治疗方案,并总结了当前的研究和未来方向。

Rutka教授论文截图
据悉,Rutka教授在小儿脑瘤综合治疗以及小儿癫痫的激光间质热疗(LITT)治疗方面尤为擅长,Rutka教授及其带领的SickKids小儿神经外科手术团队,对儿童癫痫的外科手术治疗LITT等方面积累了诸多经验,发表了许多著作。他曾担任国际神经外科学院主席、美国神经外科学院主席、美国神经外科医师协会主席。凭着其在神经外科领域的贡献,他曾获得2016年“加拿大勋章”、2006年“国际微笑勋章”,2019年他还被美国神经外科医师协会(AANS)授予“库欣勋章”(Cushing Medal)。
DIPG临床表现和诊断
DIPG患者可表现为多种神经系统症状,这也取决于肿瘤的生长位置。超过50%的患者有颅神经麻痹(面部不对称和复视等),长束征(反射亢进、上行巴宾斯基征)和小脑体征(共济失调、辨距障碍)存在。这三个经常出现的临床特征被称为“经典三联征”,应引起对该诊断的临床怀疑,促使适当的诊断成像。
鉴于DIPG进展迅速,儿童通常会在一个月或更短时间内突发症状,应引起临床注意。颅神经VI和VII较常受影响和特定的功能障碍是DIPG的重要特征。此外,虽然在诊断时不到10%的患者中观察到颅内压升高的梗阻性脑积水,这种情况通常出现在DIPG终末期。
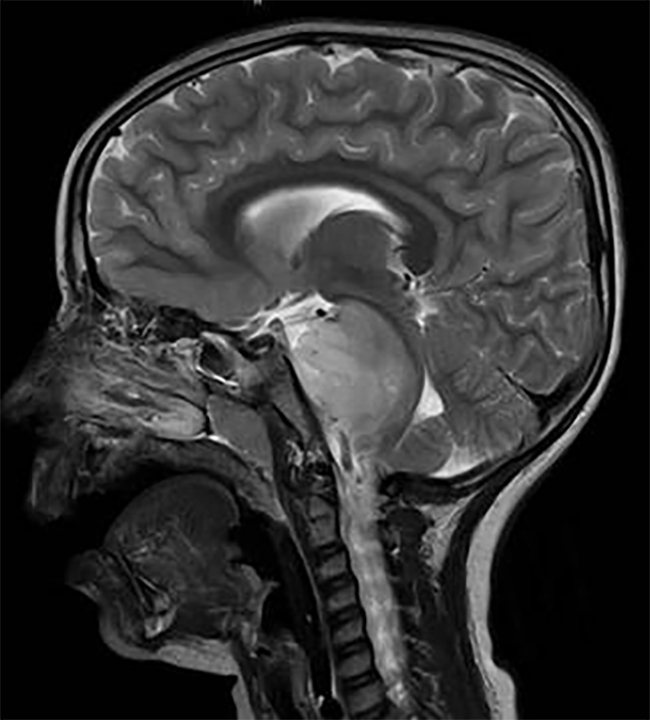
磁共振成像(MRI)是DIPG诊断的选择方式,有时也可以使用计算机断层扫描(CT)。对于这些肿瘤的浸润性质,DIPG显示T1低信号,边界不清,T2加权图像上的高信号——通常没有对比度增强(图1)。做增强扫描可能对确认诊断或排除其他病变有价值。在影像学上,肿瘤重要位于脑桥的中心,并且在出现时可以占据其轴向直径的50%以上,通常包绕基底动脉。虽然DIPG沿纤维束浸润性和弥漫性生长邻近的位置,如丘脑和小脑,它们很少转移到远处。
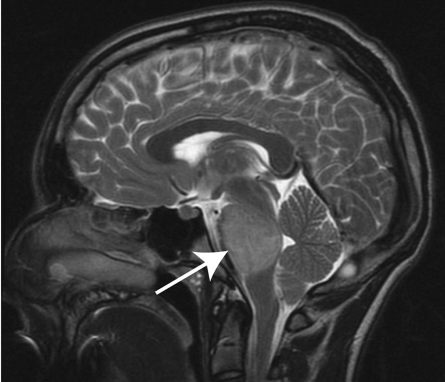
图1:T2加权矢状面(DIPG)儿童脑部MRI
随着分子诊断学在DIPG中的作用越来越大,脑干活检有助于推动该领域向前发展。多项研究表明,活检可以顺利地进行并且许多中心已经开始使用立体定向活检作为标准做法,努力加强诊断和支持基础科学研究,并应用于组织学和分子数据的临床试验。
Rutka教授所在的加拿大SickKids儿童医院也是这种诊断方法的倡导者,它可以合适获取肿瘤组织,从而深入了解这种异质性疾病的分子亚型,进而为医生进行儿童DIPG诊断、治疗提供重要参考。
鉴别诊断考虑包括非恶性脑干实体瘤,包括低级别胶质瘤、原始神经外胚层肿瘤(PNET)、血管畸形、脑炎实质病变、囊肿和脱髓鞘疾病。当通过活组织检查获得组织时,可以通过组织学检查,辅以分子检测来确诊。显微镜下,DIPG通常显示高级别星形细胞组织学,发现有丝分裂增加活性、微血管增殖或坏死(图2)。
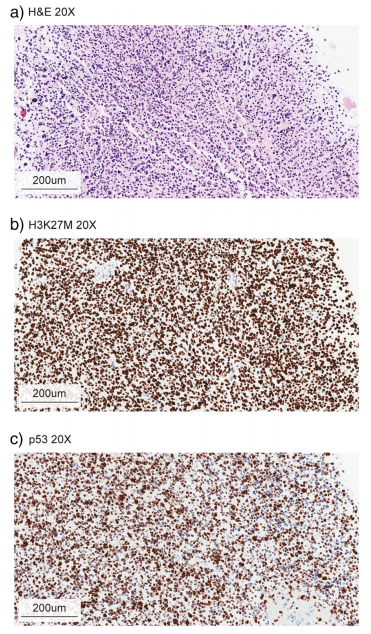
图2:DIPG组织学病理图
除了典型的胶质瘤免疫组化组,如GFAP,ATRX、p53、神经微丝蛋白、ki-67免疫染色、靶向抗体H3K27M、BRAF-V600E和IDH1-R132H的抗体可以应用(图2 b和c),各种分子病理学方法,包括新一代测序和DNA微阵列用于分子水平上确认是否存在H3突变和根据其预后确定受影响的组蛋白同种型差异。虽然治疗相关的突变例如BRAF-V600E在DIPG肿瘤中少见,通常会为了靶向治疗(如达拉菲尼或维莫菲尼)的可行性而试图进行上述分子基因评估。
分子特征和亚群
DIPGs可以细分为3个不同的分子亚群:H3K27M、MYCN和静默型。先前的研究表明,虽然DIPG与幕上高级别胶质瘤有相似之处,它仍然是一个具有独特基因和分子改变的独特实体。例如,近80%的DIPG存在H3组蛋白突变,而只有35%的儿童非脑干高级别胶质瘤具有H3突变。
尽管在DIPG中已经确定具有多种标志性突变的疾病,但应该注意的是肿瘤内和肿瘤间的异质性已被记录在此疾病。大多数DIPG中都存在组蛋白突变,并且这些突变的识别导致了我们的重点研究和临床治疗的模式转变。组蛋白突变H3K27M导致在H3.1和H3.3亚型中分别由基因HIST1H3B和H3F3A编码的赖氨酸被蛋氨酸替代。这种突变通过控制PRC2导致组蛋白三甲基化的丧失,产生表观基因沉默。然而,尽管使用活体小鼠模型进行了广泛的建模研究,H3K27M在肿瘤起始中的确切作用仍然难以捉摸。
H3.1和H3.3中的组蛋白突变存在细微差别,是对于生存、亚型和临床结果而言。H3.1组蛋白突变往往与略微提高生存率并减少转移有关。总体而言,与其他H3野生型相比,无论亚型如何,H3K27M与较差的预后结果相关。已在大约30%的DIPG肿瘤中发现ACVR1突变。之前的研究表明ACVR1突变联合其他分子突变促进肿瘤的早期进展,并在治疗靶点上显示了希望。TP53突变在大约22-40%的DIPG中发现,常与PDGFR扩增同时发生。TP53突变联合H3.3K27M和典型的PPM1D突变已被证明通过影响表观遗传调控来使肿瘤细胞逃避细胞死亡和衰老。
PDGFRA是较常见的扩增,存在于大约三分之一的高级胶质瘤中,并与RTK-RAS-PI3K-Akt信号传导通路有关。PDGFRA通过在各种磷酸酪氨酸结构域中的磷酸化作用引起PI3K和MAPK信号通路的激活。PDGFRA扩增与组蛋白H3.3突变共分离,并且无论组织学分型如何,肿瘤在临床上都具有侵袭性特点。除了PDGFRA,PIK3R1和PIK3CA也是PI3K通路的驱动因素,并已被认为会促进DIPG的侵袭性表型的作用。这些突变被描述为在H3.3K27M中的必要搭档并在DIPG的克隆群体中报道。然后,MYC和MYCN突变存在于DIPG中并发挥增强基因组范围内基因表达的转录调节因子的作用。进一步研究显示,这种疾病的基因组组观和生物学基础对更好地定义重要的致癌驱动因素或通路和随后的可执行的靶点而言是明智的。
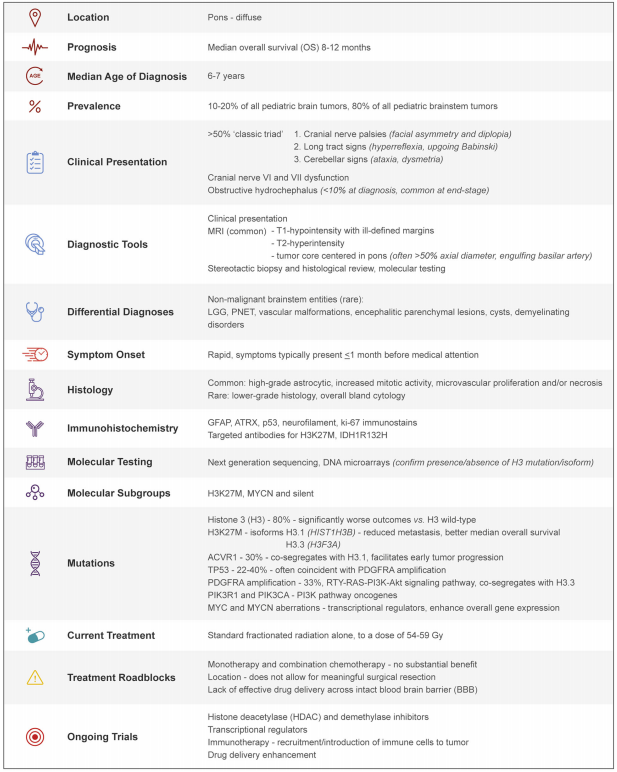
表1:DIPG临床病理特征总结
DIPG治疗标准
目前DIPG的治疗标准包括:辐射剂量为54-59Gy的单独标准分次放疗,因为任何有意义的手术切除机会都受DIPG原发主要部位的限制。此外,许多治疗方案,包括单药疗法和联合化疗迄今为止没有产生实质性的优势。然而,免疫疗法领域的进展已经确定抗GD2嵌合抗原受体(CAR)T细胞疗法的可行性,这可能会有潜在的疗效。这些有限的选择强调对新的治疗方法的需求。在此,我们介绍了D IPG治疗的可行性目标和合适治疗中常见的问题。
DIPG中的多种致癌驱动因素和体细胞突变导致肿瘤快速形成和较差的预后。正如我们之前提到的,较常见的突变包括组蛋白13的27位上赖氨酸替代蛋氨酸,是组蛋白3.1和3.3,这些都通过野生型相对物与较差的预后有关。DIPG倾向于在H3K27M中发生体细胞突变和/或H3K27三甲基的整体丢失;这同样被认为此病致癌因素之一。H3K27M的存在导致各种下游染色质重塑级联,表观遗传沉默以及各种基因和通路的激活。因此,明确这些突变及发现随后的次级突变开启了通向可治疗靶点之门,例如:组蛋白脱乙酰酶(HDAC)和去甲基化酶控制剂——其中一些显示出有希望的结果。研究还发现在临床前模型中,通过溴区蛋白的激活来靶向转录调节因子是合适的。虽然很多DIPG随着组蛋白突变发生,许多可靶向定位的次级突变也已被确定在肿瘤发生中发挥作用。
以前,关于DIPG研究和靶点识别的较常见问题之一是缺乏可获得的肿瘤组织。然而,随着尸体解剖组织和活检的增加,目前可以顺利地、可重复地进行几项分子研究。因此,许多有希望的治疗靶点已被明确并且几种药物已在临床前期试验中显示合适。然而,由于缺乏通过完整的血脑屏障(BBB)的合适药物输送,临床应用与药物研发之间仍存在相当大的障碍。这或许可以解释为什么在其他神经胶质瘤中显示出疗效的药物在DIPG中失败。经过结构上的改变或物理损害BBB来好转药物输送将会成为对可以转化为临床的新疗法至关重要的一环。
然后,肿瘤微环境是决定治疗(是免疫治疗)时要考虑的肿瘤的重要组成部分。近期的研究有结论是DIPGs具有非炎症性肿瘤微环境。然而,关于DIPG肿瘤是否含有肿瘤相关巨噬细胞,由于结果存在矛盾,尚未得到充分研究。这些结果表明DIPGs没有增加巨噬细胞浸润或DIPGs增加巨噬细胞浸润但不分泌炎性细胞因子。也就是说,大多数研究表明,DIPG中没有T细胞浸润,因此免疫治疗方法应侧重于将免疫细胞募集至肿瘤中而发挥治疗效果。
DIPG新的治疗途径
DIPG有多种对未来有希望的治疗途径。这些包括靶向疗法、基因疗法和免疫疗法。在这里,我们简要描述这些治疗方式,并突出它们的现状及临床意义。
自从DIPG靶向治疗的发展以来,已启动约250项临床试验对抗疾病中的不同生物学途径。较常扩增的基因之一是PDGFRA,它存在于10%的DIPG中。因此,PDGFRA是DIPG治疗中有针对性的基因之一。然而,靶向PDGFRA的药物如伊马替尼和达沙替尼在临床实验中表现出相当差的抗肿瘤作用。DIPG中靶向的另一个基因是EGFR,这也已被证明在小儿脑肿瘤中过度表达。抗-EGFR的临床试验包括尼妥珠单抗、吉非替尼和厄洛替尼在DIPG患者的小型子集中显示的效果。其他试验有使用PARP1控制剂(奥拉帕尼、尼拉帕尼、维利帕尼),CDK4/CDK6控制剂(PD-0332991)、WEE1激酶控制剂(MK1775)和血管生成控制剂(贝伐单抗)。尽管经过各种临床试验尝试,这些都没有在DIPG显示出显着的功效。这些临床试验中的限速步骤之一是关于这些药物是否穿过BBB的研究仍有许多未解之谜。
近期,大量证据表明表观遗传改变合并基因突变是肿瘤发生的原因。使用JMJD3控制剂的研究如帕比司他和GSK-4等分别靶向组蛋白去乙酰化酶和去甲基化酶,显示出有希望的结果,目前已经进入作为单一和联合药物的临床试验(图3a和b)。H3K27me3水平降低导致了针对染色质重塑的独特策略。zeste同源物2(EZH2)的增强子是一种H3K27-甲基化酶并被发现在DIPG的H3K27M突变体中显示高表达。然而,用EZH2控制剂的治疗方案EPZ6438在GBM和DIPG细胞系中几乎没有结果。相比之下,tazemetosta(他泽司他)(图3c)也是一种EZH2控制剂,产生了明显更好的结果,尽管这可能是由于样本选择偏差。
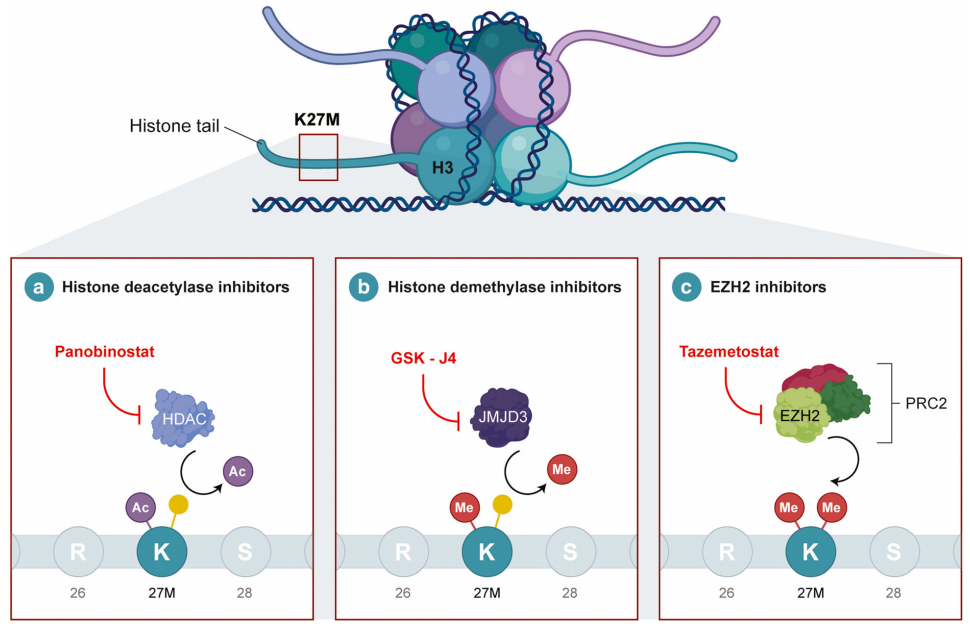
图3:儿科DIPG的药物靶点和治疗。DIPG的特征在于组蛋白H3中发生的K27M突变
另一方面,研究还针对负责H3K27去甲基化的酶,如JMJD3。BET家族蛋白等转录调节因子也被研究作为脑肿瘤治疗的靶点。已发现JQ1是一种组蛋白结合控制剂,可与溴结构域结合并取代BRD4融合癌基因蛋白随后导致细胞周期停滞和细胞凋亡(图4a)。此外,干扰转录的替代方法也已合适,例如用THZ1控制CDK7(图4b)。除了这些分子靶点,其他几个二次突变尚未被作为可行的治疗进行研究(表2)。
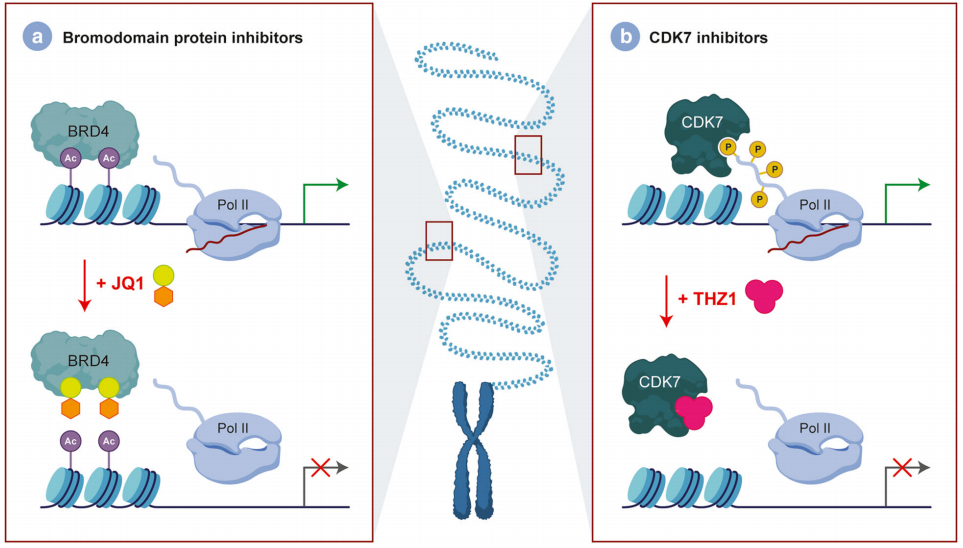
图4:DIPG中可靶向的转录依赖性
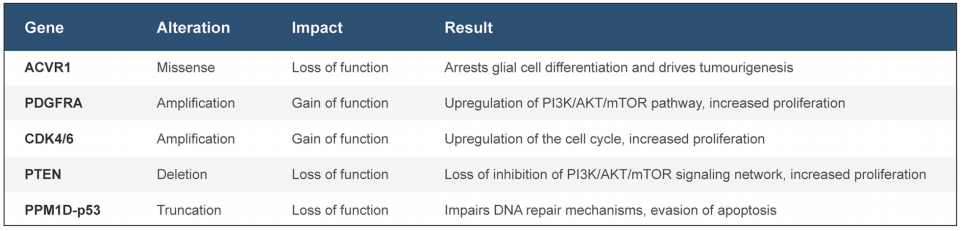
表2:儿科DIPG中潜在的可靶向二次突变
新的证据已将表观遗传学和代谢组学与可塑性和脑肿瘤的瘤内异质性联系起来。特别是在DIPG中,近期研究发现代谢重编程促进H3.3K27M DIPG的发病机制,主要通过利用α-酮戊二酸来维持低H3K27me3的优先表观遗传状态。此外,他们还表明H3.3K27M细胞在使用葡萄糖或谷氨酰胺来调节总体H3K27me3时依赖一种或两种途径显示出瘤内异质性。结果,总体H3K27me3的代谢调节导致对谷氨酸脱氢酶、己糖激酶2和野生型异柠檬酸脱氢酶1(IDH1)的异质依赖性,这些可能是具有治疗作用的靶点。利用DIPG中的这些代谢和表观遗传易损性应该是对未来治疗策略的优先级环节。
免疫疗法正在迅速确立自己在癌症治疗中的支柱地位,近期的研究表明了它在脑肿瘤中的潜能。特别是,一个抗GD2 CAR-T研究已在DIPG活体模型中表现出乐观的结果。然而,值得提出的是,这些GD2 CAR的使用导致了脑积水,这在一部分动物试验中是致命的,这被认为是由于肿瘤在脑脊液通路的神经解剖位置而引起。鉴于这些挑战,明确应用免疫疗法的新策略的适应症,将会要求建议把这些有希望的治疗方案推进临床试验。
结论
复杂的分子发病机制、严密的血脑BBB调控和不同的位置促成了目前DIPG的预后差。放射治疗仍然是主要的治疗方法。然而,随着对DIPG分子基因学的认识不断加深,将有越来越多的有希望的临床前研究和新技术来克服透过血脑屏障合适输送药物的局限性,我们有理由相信DIPG未来的治疗选择将越来越多、疗效越来越好。
- 文章标题:脑干胶质瘤DIPG——从未被放弃的医学难题,INC小儿神外Rutka教授如何攻克?
- 更新时间:2024-01-25 09:21:22