400-029-0925
400-029-0925髓上皮瘤是什么病?髓上皮瘤(ME)是一种少见的,高度恶性肿瘤,通常出现在婴儿和儿童早期,一半的病例发生在2岁以下的儿童。由Bailey和Cushing在1926年一次描述的髓上皮瘤被归类为原始神经外胚层肿瘤,其特征是形成类似胚胎神经管的假层神经上皮瘤结构典型的髓上皮瘤发生在眼内区域或沿神经轴,而在眼眶或盆腔等周围部位发现的髓上皮瘤很少报道。肿瘤的位置是影响生存的重要预后因素位于眼内区域的肿瘤通常是良性的,边界清楚,可以完全切除,而中枢神经系统(CNS) 髓上皮瘤具有高度侵袭性,据报道中位生存期为5个月。
病例报告
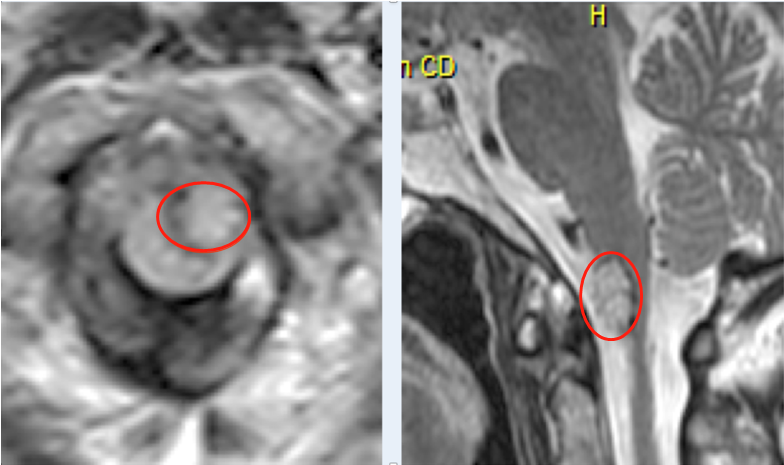
1例2岁女童有2- 3周的急症病史并伴有严重腹痛。磁共振成像显示有一个较大的骶前肿块,长度为6.3×5.0×5.7 cm,并延伸至骶神经孔S4和S5处。未见转移(图1)。活检显示乳头状肿瘤伴假层柱状细胞和深染小圆细胞肿瘤,伴玫瑰花结、假玫瑰花结和血管周围玫瑰花结。显微镜方面也显示出坏死的出血区域和神经元和星形细胞分化的证据。肿瘤中缺乏间充质或内胚层成分排除了畸胎瘤的诊断。

图1:A,T2磁共振成像在诊断时显示骶前肿块。B, T1, gad后磁共振成像显示化疗后骶骨前肿块
病理报告与骶前髓上皮瘤一致,患者按照CCG 99703协议进行治疗患者接受顺铂、长春新碱、环磷酰胺、依托泊苷诱导方案,卡铂、硫替帕巩固化疗2个周期,并辅以自体干细胞挽救。经1个疗程诱导化疗后,症状迅速缓解。
化疗5个月后,对照磁共振成像显示病变明显缩小(4.6×2.6×2.0 cm),允许手术完全切除。为了实现完全切除,切除尾骨远端一小块。术后病程无明显变化,病理报告显示无存活肿瘤的证据。术后1个月,患者接受了较后一个周期(三个周期)的巩固治疗(卡铂+硫替帕+自体干细胞挽救)。术后选择每日雷帕霉素的辅助维持治疗,不给予辅助放疗。然而,由于耐受性问题,在家长的要求下停用了雷帕霉素。在确诊后5年的较后一次随访中,患者生存良好,无长期性后遗症或肿瘤复发迹象。
案例分析
髓上皮瘤是一种少见的恶性肿瘤,通常出现在眼内区域或中枢神经系统,它们通常在5岁以下儿童的大脑半球发现,颅外髓上皮瘤的临床病例很少。
考虑到我们的患者和诊断时肿瘤的大小,我们决定继续化疗作为一道治疗,以诱导肿瘤大小的大幅减小。事实上,我们的主要目标是防止激进手术造成的神经损伤。此外,考虑到肿瘤的大小及其向神经孔的延伸,全切除是困难的。虽然我们的方法是成功的,但由于不同的肿瘤生物学或抗肿瘤药物的不同选择,De Pasquale等人报道的病例中没有相同的策略。
结论
本报告补充了现有的关于这种少见病的文献,并建议在这种少见的情况下,使用高剂量和高剂量的化疗来保留辐射可以获得良好的结果。
相关参考资料来源:DOI:10.1097/MPH.0000000000001460
- 文章标题:髓上皮瘤是什么病?
- 更新时间:2021-04-25 09:51:51