400-029-0925
400-029-0925髓母细胞瘤包括一系列发生在后颅窝的肿瘤亚群。这种恶性肿瘤主要发生在婴儿和儿童,中位年龄为8岁,男女比例为1.5:1。它约占全部儿童脑肿瘤的20%,是较常见的儿童脑肿瘤。然而,10-25%的髓母细胞瘤(MBs)发生在青少年和成人中,在40岁后很少发生。每年的发病率估计约为每百万人中有10名儿童和0.5名成人。导致诊断的症状主要包括嗜睡、呕吐、头痛和躯干性共济失调。
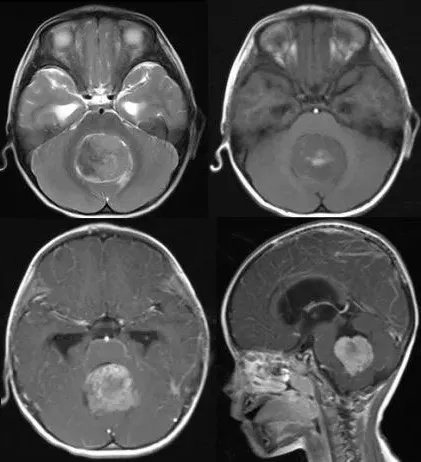
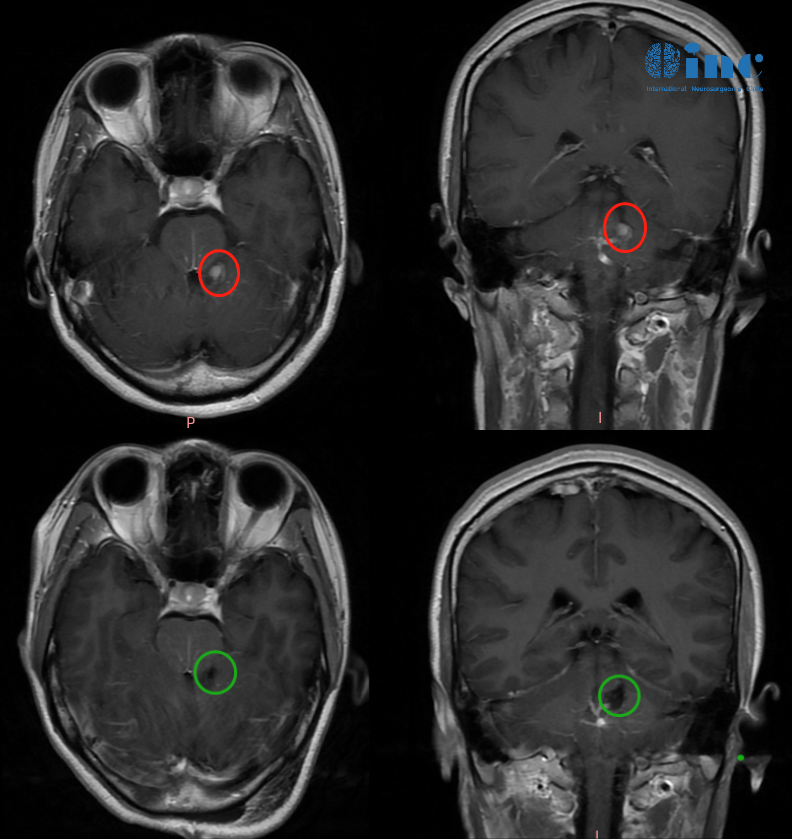
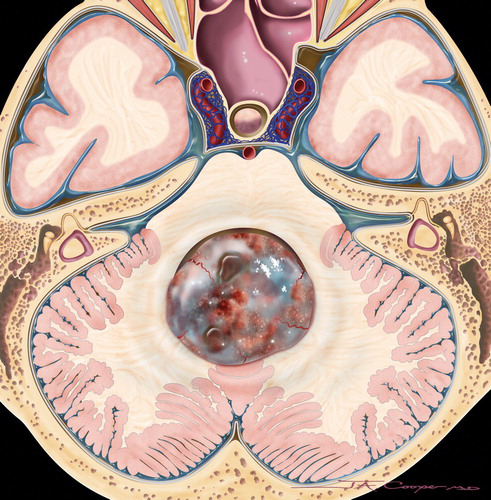
髓母细胞瘤典型影像
在当代治疗中,髓母细胞瘤患者的5年生存率高于70%。因此,与其他神经上皮性肿瘤,如弥漫性内在桥脑胶质瘤(WHO II-IV)或胶质母细胞瘤(WHO IV)相比,大多数髓母细胞瘤患者的预后是明显是有利的。髓母细胞瘤患者的临床异质性以及类似分级胶质瘤预后的差异说明了WHO实际分级在预后和评估价值方面的局限性。标准更好地评估髓母细胞瘤的行为和对治疗的反应是必要的。
目前髓母细胞瘤治疗方案建议较大限度的顺利肿瘤切除,然后进行化疗,对于3岁以上的患者,给予额外的放疗。髓母细胞瘤患者的治疗反应和总生存率有差异,即使在组织病理学和疾病程度相当的病例中也是如此。通过大规模基因组分析,允许将髓母细胞瘤分为至少4个具有独特分子谱的亚组(WNT、SHH、组3和组4),并将这些数据与人口统计学参数和临床结果相关联。WNT髓母细胞瘤瘤患者的“治愈率”超过90%,而三组的治愈率为40-60%,这说明需要进一步改进患者特异性治疗方案。分子分型有前景提供一种额外的手段,通过未来的治疗方案的发展。
在INC国际神经外科医生集团旗下国际神经外科顾问团成员、国际神经外科杂志《Journal of Neurosurgery》主编、加拿大多伦多大学儿童医院Sickkids大外科主任及脑瘤研究中心主席James T.Rutka教授的《Medulloblastoma:Tumor Biology and Relevance to Treatment and Prognosis Paradigm》论文中,综述了目前针对儿童髓母细胞瘤的生物学分型、诊断、治疗及预后因素的分析。


髓母细胞瘤的分型及治疗预后
分子亚组反映了临床结果,2006年汤普森等人提出了一批研究表明,髓母细胞瘤可以根据相似的基因组表达谱。2010年诺斯科特等人在转录组学和DNA拷贝数改变的基础上进一步定义了基因组特征,并将分子细分组整合到患者的人口统计学和临床结果中。在接下来的5年里,一些实验室验证了包括1000多名患者在内的队列组织样本。这一累积数据提供了关于染色体改变和突变谱的结果。
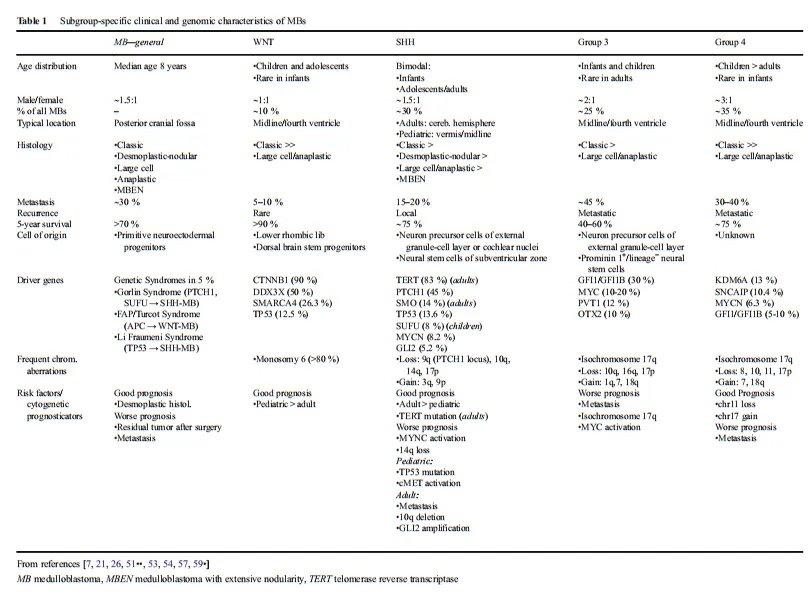
2010年,由国际卫生组织(WHO)组织的专家团一次将髓母细胞瘤通过分子标记物分成四个分子亚型:WNT活化型、SHH活化型、Group 3型和Group 4型。WNT和SHH肿瘤分别通过确定导致WNT和SHH通路上调的突变来标记。3组和4组的潜在基因驱动因素还不太清楚。到目前为止,还没有确定决定性的路径改变;因此,使用了通用名称。
当将临床过程与遗传分型相关联时,需考虑一个重要的方面:分子亚组的实际数据已从根据临床分层方案治疗的患者中获得,而没有考虑特定的遗传特征。因此,目前尚不清楚观察到的临床结果差异在多大水平上可归因于亚组的生物学特性(预后因素)或基于分子的肿瘤对当代治疗的易感性(评估因素)。未来临床试验的挑战是逐步探索治疗方法,整合髓母细胞瘤基因谱的进化数据的治疗方法。
WNT子组
WNT亚群的特征是改变导致WNT通路的活性的增加。在全部的亚型中,它具有较一致的遗传畸变模式,几乎全部的肿瘤都具有经典的组织学。WNT仅占全部MB肿瘤的10%,是较少见的,但在全部MB亚型中预后较好。在当代的治疗方案中,5年生存率超过90%,前提是已经进行了总的全切除术,并且没有转移性。这些肿瘤通常发生在年龄较大的儿童和青少年中,性别比例分布均匀。3岁婴儿<的表现少见。沿脑脊液途径的转移相对少见,占5-10%。WNT髓母细胞瘤通常位于四脑室中线区域,肿瘤常表现为脑干浸润。
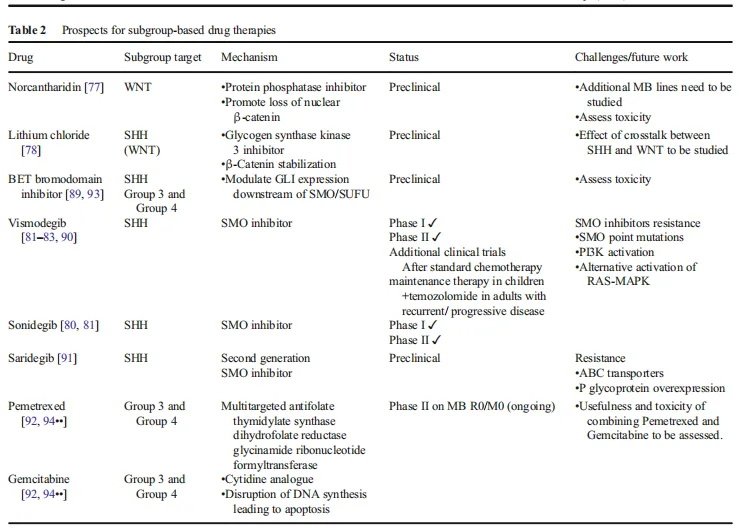
▪治疗方式:WNT信号通路早在30多年前就一次被描述过,但旨在调节或控制该通路上的点的药物直到近期才进入临床试验。到目前为止,这些药物都没有被批准用于癌症治疗,也没有临床试验被提倡用于治疗的药物。控制WNT信号通路的一个关键因素是SHH和WNT通路之间的串扰,这在不同程度上可以被描述为协同或相反的。例如,在一个临床前模型中,蛋白磷酸酶控制剂降甘肽已被证明可以改变WNT信号通路和通过促进核β-连环蛋白的丢失来阻止髓母细胞瘤的生长。另一方面,Zinke等人。结果表明,糖原合酶激酶3控制剂氯化锂对β-连环蛋白的稳定降低了SHH驱动的MBs的生长。鉴于WNT髓母细胞瘤具有较高的治愈率和良好的结果,在不久的将来,临床研究将更多地关注于WNTMB标准治疗的改进。
SHH子组
SHH信号通路的突变可能会在约30%的MBs中引发肿瘤的形成(Northcott 2012)。男女比例为1:1,发病率呈双峰模式。3岁以下或16岁以上的髓母细胞瘤患者中有三分之二患有SHH-MB。在15-20%的中存在转移性传播。总体5年生存率约为75%,因此,与WNT和组3亚型相比,它表现为中等预后。与WNT-MBs相比,其组织学和分子特征以及临床结果表现出更多的变异性。在全部亚组中,只有SHH-MBs可能表现出全部五种主要的组织病理学亚型,尽管经典的和结缔组织增生性亚型是较常见的。几乎全部显示结缔组织增生或结节性组织学的髓母细胞瘤都属于SHH亚组。导致SHH活性增加的较常见的基因突变包括PTCH1(45%)、SMO(14%)和SUFU(8%)。
在儿童SHH髓母细胞瘤中,肿瘤多发生在中线(蚓部),而在成人中,肿瘤表现多见于小脑半球。这种年龄倾向可能归因于不同的肿瘤起始驱动突变。在儿童和儿童和成人(>16岁)患者队列中,基因谱的变异性对生存率有决定性的影响。成人SHH MBs往往比儿童队列[50]有更好的总生存期。TP53突变在MBs中几乎只在WNTand SHH肿瘤中发现,它可能是儿童和青少年中SHH MBs较重要的危险因素。
▪治疗方式:目前,MB治疗的分子靶点较能研究异常的SHH通路。调节SHH信号的方法主要集中在SMO控制上。SMO控制剂如视觉药物和超声波已经被FDA批准用于基底细胞癌的治疗。在一份病例报告和一项I期研究显示,视觉治疗后SHH髓母细胞瘤的肿瘤生长明显减少(但不是持续减少)后,2015年进行了一项更大的II期研究,包括43例复发MBs患者(12例SHH肿瘤)。41%的SHH患者获得了无进展生存期和疾病稳定。4例SHHMB患者持续(>8周)病变完全或部分消失,而WNT、3组或4组肿瘤患者均未出现反应。研究发现,对视觉敏感的SHH MB患者的PTCH1功能突变缺失,而治疗失败的SHH MB肿瘤在SMO下游的SUFU和GLI2中存在基因畸变。
这些发现证实了先前来自异种移植模型的数据,即SMO控制剂仅在含有SMO上游突变的SHH-MBs子集中合适。此外,已存在的或获得性的耐药性可能会限制SMO控制剂的长期疗效。SMO的点突变、PI3K信号通路的增加或RAS-MAPK通路逃避SHH通路的替代激活导致耐药性。将SMO拮抗剂与PI3K阻滞剂或GLI控制剂联合使用可能是延缓耐药性发展的一种策略。BET溴域控制剂可调节SMO和SUFU下游的GLI表达,因此也被认为是很有前途的药物。
3组
约25%的MBs占组3亚组。这类患者的5年生存率约为50%,是全部亚型中结果较差的。3组MBs几乎从未在成人中出现过;大多数患者为婴儿和儿童,男女比例为2:1。大多数3组MBs具有典型的组织学特征;然而,这一组包含了较高的LCA组织学比例(40%),特别是在婴儿中。转移性表现的发生率高达45%,并伴有较差的预后。与4组一样,目前还没有发现明显的主要信号通路的改变;因此,诊断主要是基于转录谱聚类。
这些肿瘤表现出一个不平衡的基因组,具有各种模式的染色体和遗传畸变。高达30%的基因显示增强子区域的基因组重排,导致致癌基因GFI1和GFI1B的过度激活,这是迄今为止发现的3组中较普遍的遗传改变。此外,该亚组中MYC(10-20%)、PVT1(12%)和OTX2(10%)的扩增率相对较高。同位染色体17q发生在全部3组MBs的25%左右,它被认为是较差的预后的一个强有力的预后因子。
4组
4组髓母细胞瘤是较常见的亚组,总占全部髓母细胞瘤的35%。这种肿瘤在婴儿中相对少见,但它占了近50%的儿童髓母细胞瘤和全部成人MBs的25%。男性受到的影响通常是女性的两到三倍。经过标准治疗后,患者预后中等,与shh组相当,尽管软脑膜转移明显更常见(30-40%)。然而,与至少非成人SHHMBs相比,转移状态似乎是4组MBs更糟糕的结局的一个更强的评估因子。组织形态学模式通常为典型或LCA外观。在大约80%的病例中,较常见的基因改变是等色一些17q,尽管尚未被证明对3组有预后作用。较常见的突变影响KDM6A基因(13%),该基因位于X染色体上,编码一个组蛋白去甲基化酶(H3K27)。这种与X染色体相关的突变可能导致了该亚群的男性优势。与SHHMBs类似,在10%的<中发现了癌基因MYNC的扩增,其作为预后标志物的影响尚不确定。
▪治疗方式:这两组之间缺乏明确的基因驱动因素和分子谱的重叠,使靶标治疗候选药物的研究变得复杂。对3组髓母细胞瘤的临床前研究大多采用MYC驱动的动物模型。应用高通量对过表达MYC的MB细胞进行筛选,fda批准的两种药物培美曲塞和吉西他滨可控制增殖。这些药物在小鼠同种异体移植和异种移植显示肿瘤生长减少。BET溴域蛋白已被证明在各种癌症中控制myc相关通路的活性,近期有临床前证据表明myc驱动的MBs存在反应。需考虑的是,尽管MYC是一种普遍的特征,但仅在约10-20%的患者样本中发现了MYC表达水平的升高。此外,关于MYC活性增加的预后价值也存在争议。
总结
在过去的5年里,我们对所观察到的髓母细胞瘤临床异质性的分子模式的理解增加。即将进行的临床试验将亚组特异性基因标记纳入传统的临床风险分层方案,将逐步完善和定义分子谱的预后和评估价值,以支持预防和治疗干预。基因驱动因子定位的进展有使靶向治疗成为可能的前景,这将增加化疗设备。预计根据髓母细胞瘤的分子背景进行选择性调整的辅助治疗模式将好转肿瘤控制,降低治疗诱导的发病率。
儿童髓母细胞瘤是儿童中枢神经系统肿瘤的高发肿瘤,其恶性程度高,易出现播散转移,在及时标准的治疗模式下,大部分可以取得良好的预后。对于高危和复发难治的髓母细胞瘤,目前无标准治疗方案,需联合放疗、化疗、免疫、靶向等综合治疗手段。基于分子分型和遗传学特点的个性化治疗是未来探索的方向。
INC加拿大James T.Rutka教授表示,在过去的20多年里,我们越来越熟悉小儿髓母细胞瘤特有的患者症状群、特定的神经影像学特征以及标准化的治疗方法。虽然手术切除及治疗仍然面临着较大挑战,其术前细致的手术评估、合理的手术方案的确定和有经验的手术团队至关重要。

据悉,Rutka教授曾连续三年任职国际神经外科学院院长,同时还是美洲神经外科学院前院长、美洲神经外科医师协会主席,如今作为国际神经外科杂志《Journal of Neurosurgery》主编,其自身发表了超过500多篇的文章,著有神经外科专著多本,在临床上的研究方向以颅内肿瘤为主,对胶质瘤、纤维瘤、颅咽管瘤、室管膜瘤、松果体区肿瘤等具有多年的临床经验,擅长儿童脑瘤的分子分型研究和综合治疗以及小儿癫痫的外科治疗,包括激光间质热疗(LITT)、清醒开颅术等显微外科手术。
- 文章标题:5年生存率超过90%,不同分型髓母细胞瘤预后治疗有何区别?
- 更新时间:2024-06-23 16:46:05