400-029-0925
400-029-0925脊髓脊膜膨出是一种影响中枢神经系统的畸形病变。早期的治疗对新生儿的存活和功能障碍有着深远的影响,因为功能障碍将会伴随他们一生,所以这些存在复杂畸形的患儿需要而的医生熟悉他们的问题。脊髓囊样膨出并不常见,在新生儿发病的早期与病灶相关症状较少,但需要长时间的神经外科治疗。
脊髓脊膜膨出是在人类胚胎形成的18~27天时,由于初级神经胚形成的异常所致。位于神经基板中间的神经沟是中央管的残迹。脊髓神经根由神经基板的前面发出,腹根排列在内侧而背根位于外侧。融合缺陷的硬脊膜在筋膜的外侧,功能性的神经组织可能存在于神经基板的尾部或是在从神经基板上发出的神经根上。

大部分的脊髓脊膜膨出(85%)发生在胸腰部末端或更远端,10%在胸部,其余在颈部。在颈部的脊髓脊膜膨出常与脊髓囊样膨出相似,不伴有 ChiariⅡ型畸形和脑积水。
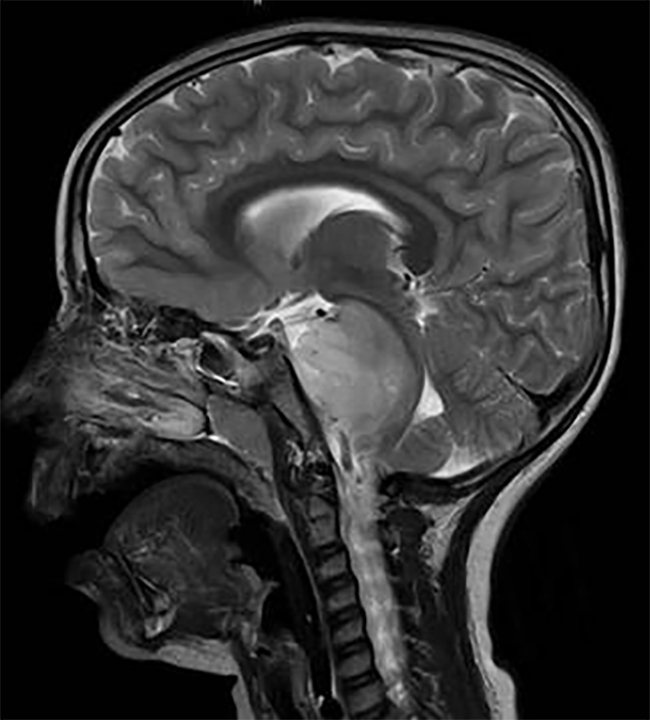
几乎全部的脊髓脊膜膨出患儿都伴有 ChiariⅡ型畸形,以及一系列影响中枢神经系统的异常病变。其中,脑干缺陷包括:延髓扭结、顶盖变尖及内部神经核异常。幕上异常包括:部分或是整个胼胝体发育不全、多小脑回、较大的丘脑间粘合以及灰质异位。神经中胚层的发育也会受到影响,出现后颅窝狭小、斜坡变短、小脑幕和窦汇位置降低,切迹变宽,枕骨骨孔增大, Lüchenschädel病或称为顶骨内陷,是指脊髓发育不良的婴儿其中胚层的颅骨发育异常。这些病变不是颅内压增高所致。通常到1岁时逐渐好转。
大部分的脊髓脊膜膨出患儿(80%~90%)伴有脑积水并需要治疗。脑积水是由于阻塞性或交通性因素引起。有40%~80%的脊柱裂患者发生脊髓空洞症,但通常空洞不再发展。脊柱裂患儿的神经系统变性可能由脑积水或 ChiariⅡ型畸形造成,也可能是由于脊髓空洞症或是组织粘连。但神经系统变性较常见的原因是由于分流失败导致的脑积水。
病因学
脊柱裂有多种病因,因为在某些种族群体中该病有着很高的发病率所以被认为可能有基因遗传的倾向。营养物质的缺乏,是叶酸和锌的缺乏,在20世纪80年代中期,已被证实是该病的病因。
脊髓脊膜膨出预后好吗?
脊髓脊膜膨出患儿的2年存活率是95%,10%-15%的脊柱裂患儿死于六岁之前,甚至是在的治疗中死亡。而需要治疗的脑积水发生率是90%-大概率。
运动平面决定了患儿的行走状态,但运动平面不与脊髓脊膜膨出的解剖平面相符合。产前解剖平面可用超声检查准确测量,能准确地评估患儿在3-4岁时的运动功能,这种方法优于通过新生儿检查进行评估。L3神经的功能是使人站立,L4与L5的功能是使人移动。在10岁前:大约60%的脊柱裂患儿依靠或是不依靠辅助工具可以在社区内活动;26%不能运动;15%可以在家庭内活动。在青春期时,能在社区内活动的患者的比例下降了17%。在青少年时许多脊柱裂患者需要通过各种设备提高运动辅助的水平。有很少一部分(6%-17%)的患儿能控制小便。大部分(85%)的患儿需要不断更换导尿管和药物治疗,86%的患儿能控制大便。
大多数(75%-80%)脊柱裂患儿经过进一步治疗脑积水和感染后,智力可以是正常的。在一些调查研究中发现:高位脊髓脊膜膨出以及严重的脑室增大与智力低下相关。如果中枢神经系统没有感染,其智商(IQ)应是正常的,但大约有1/3(31%)中枢神经系统曾经感染的患儿,其IQ也是正常的。分流术的次数是否影响IQ仍然存在争论。尽管脊柱裂患儿大多不具有复杂学习能力,但有超过一半(58%)患儿的学习能力可以达到同龄正常儿童的水平。有10%~15%的患儿因严重的认知损害而需要监护。有很少一部分(<10%)患儿生活可以自理;大部分(>80%)脊柱裂患儿需要精神护理和咨询,来帮助他们面对残疾。
骶尾部脊髓脊膜膨出与脊髓囊样膨出类似,预后很好。大部分颈部脊髓脊膜膨出的患者伴有脑积水,并且需要行分流手术。
关于您具体的预后情况,没有看到您的病历和检查资料,很难一概而论,但是你真的很棒了!
相关参考资料:Youmans Neurological Surgery
- 文章标题:脊髓脊膜膨出是什么病?预后好吗?
- 更新时间:2021-03-25 10:43:01